20 KiB
FScanpy 教程 - 完整使用指南
![]()
摘要
FScanpy 是一个专为预测 DNA 序列中程序性核糖体移码(PRF)位点而设计的 Python 包。该包集成了机器学习模型、序列特征分析和可视化功能,帮助研究人员快速定位潜在的 PRF 位点。
介绍

FScanpy 是一个专门用于预测 DNA 序列中程序性核糖体移码(PRF)位点的 Python 包。它集成了机器学习模型(梯度提升和 BiLSTM-CNN)以及 FScanR 包,提供精确的 PRF 预测。用户可以使用三种类型的数据作为输入:需要预测的完整 cDNA/mRNA 序列、疑似移码位点附近的核苷酸序列,以及物种或相关物种的肽库 blastx 结果。它预期输入序列位于 + 链上,并可与 FScanR 集成以提高准确性。
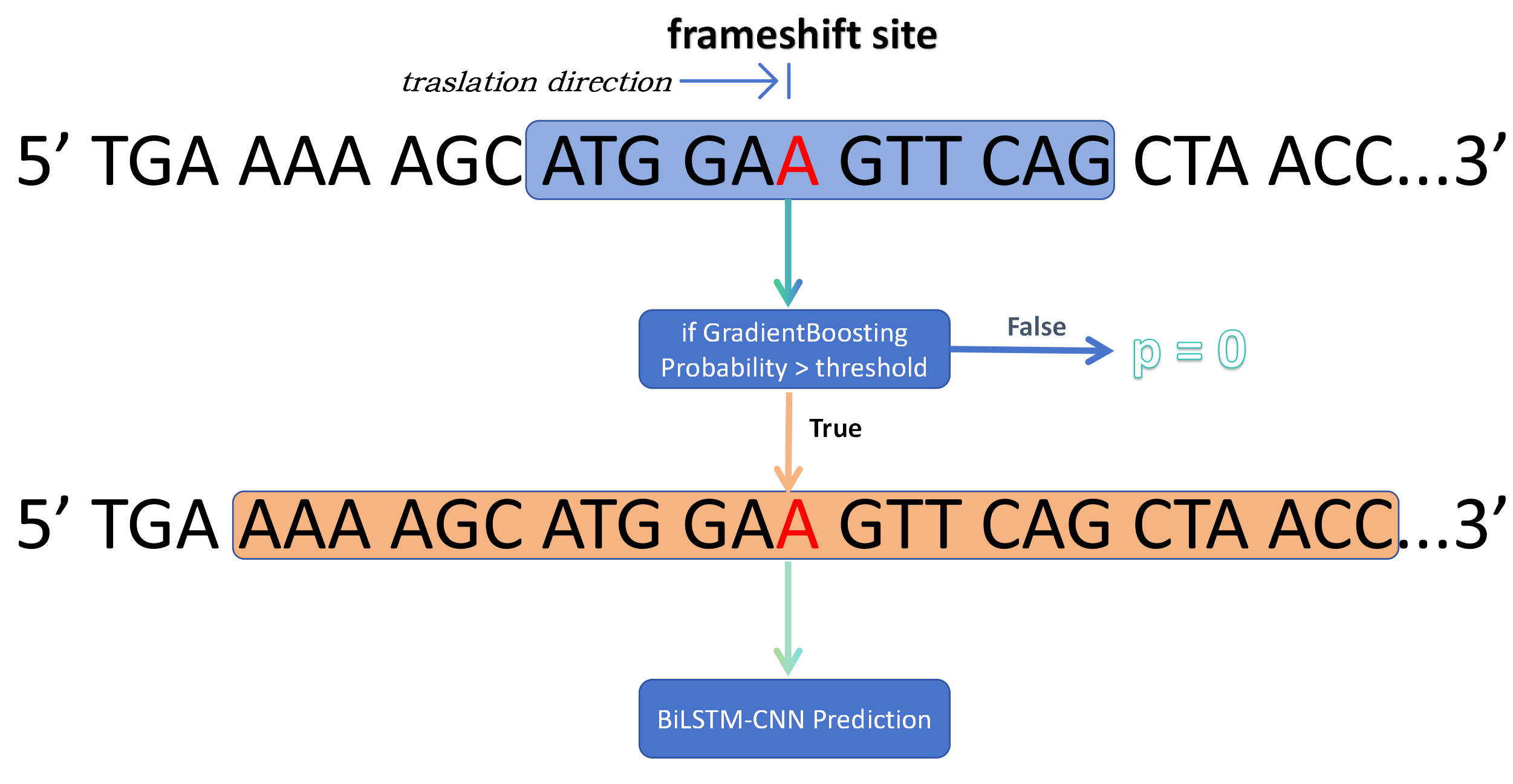
对于整个序列的预测,FScanpy 采用滑动窗口方法扫描整个序列并预测 PRF 位点。对于区域预测,它基于疑似移码位点周围 0 读码框中的 33bp 和 399bp 序列。首先,短模型(HistGradientBoosting)将预测扫描窗口内的潜在 PRF 位点。如果预测概率超过阈值,长模型(BiLSTM-CNN)将预测 399bp 序列中的 PRF 位点。然后,集成权重结合两个模型进行最终预测。
对于从 BLASTX 输出检测 PRF,FScanR 从 BLASTX 比对结果中识别潜在的 PRF 位点,获取同一查询序列的两个命中,然后利用 frameDist_cutoff、mismatch_cutoff 和 evalue_cutoff 过滤命中。最后,使用 FScanpy 预测 PRF 位点的概率。
背景
核糖体移码,也称为翻译移码或翻译重编码,是翻译过程中发生的一种生物现象,导致从单个 mRNA 产生多个独特的蛋白质。该过程可由 mRNA 的核苷酸序列程序化,有时受二级、三维 mRNA 结构影响。它主要在病毒(特别是逆转录病毒)、逆转录转座子和细菌插入元件中被描述,也在一些细胞基因中存在。
FScanpy 的主要特性包括:
- 两个预测模型的集成:
- 短模型(HistGradientBoosting):分析以潜在移码位点为中心的局部序列特征(33bp)。
- 长模型(BiLSTM-CNN):分析更广泛的序列特征(399bp)。
- 支持跨多种物种的 PRF 预测。
- 可与 FScanR 结合以提高准确性。
安装 (python>=3.7)
1. 使用 pip
pip install FScanpy
2. 从 GitHub 克隆
git clone https://github.com/.../FScanpy.git
cd FScanpy
pip install -e .
完整函数参考
1. 核心预测函数
1.1 predict_prf() - 主要预测接口
函数签名:
def predict_prf(
sequence: Union[str, List[str], None] = None,
data: Union[pd.DataFrame, None] = None,
window_size: int = 3,
short_threshold: float = 0.1,
ensemble_weight: float = 0.4,
model_dir: str = None
) -> pd.DataFrame
参数:
sequence:用于滑动窗口预测的单个或多个 DNA 序列data:DataFrame 数据,必须包含 'Long_Sequence' 或 '399bp' 列用于区域预测window_size:滑动窗口大小(默认:3,推荐:1-10)short_threshold:短模型(HistGB)概率阈值(默认:0.1,范围:0.0-1.0)ensemble_weight:短模型在集成中的权重(默认:0.4,范围:0.0-1.0)model_dir:模型目录路径(可选,如果为 None 则使用内置模型)
返回值:
pd.DataFrame:预测结果,包含以下列:Short_Probability:短模型预测概率Long_Probability:长模型预测概率Ensemble_Probability:集成预测概率(主要结果)Position:序列中的位置(滑动窗口模式)Codon:位置处的密码子(滑动窗口模式)Ensemble_Weights:权重配置信息
使用示例:
from FScanpy import predict_prf
# 1. 单序列滑动窗口预测
sequence = "ATGCGTACGTATGCGTACGTATGCGTACGT"
results = predict_prf(sequence=sequence)
# 2. 多序列预测
sequences = ["ATGCGTACGT...", "GCTATAGCAT..."]
results = predict_prf(sequence=sequences)
# 3. 自定义参数
results = predict_prf(
sequence=sequence,
window_size=1, # 扫描每个位置
short_threshold=0.2, # 更高阈值
ensemble_weight=0.3 # 3:7 比例(短:长)
)
# 4. DataFrame 区域预测
import pandas as pd
data = pd.DataFrame({
'Long_Sequence': ['ATGCGT...', 'GCTATAG...'], # 或使用 '399bp'
'sample_id': ['sample1', 'sample2']
})
results = predict_prf(data=data)
1.2 plot_prf_prediction() - 带可视化的预测
函数签名:
def plot_prf_prediction(
sequence: str,
window_size: int = 3,
short_threshold: float = 0.65,
long_threshold: float = 0.8,
ensemble_weight: float = 0.4,
title: str = None,
save_path: str = None,
figsize: tuple = (12, 8),
dpi: int = 300,
model_dir: str = None
) -> tuple
参数:
sequence:输入 DNA 序列(字符串)window_size:滑动窗口大小(默认:3)short_threshold:热图显示的短模型过滤阈值(默认:0.65)long_threshold:热图显示的长模型过滤阈值(默认:0.8)ensemble_weight:集成中短模型的权重(默认:0.4)title:图表标题(可选,如果为 None 则自动生成)save_path:保存路径(可选,如果提供则保存图表)figsize:图形大小元组(默认:(12, 8))dpi:图形分辨率(默认:300)model_dir:模型目录路径(可选)
返回值:
tuple:(prediction_results: pd.DataFrame, figure: matplotlib.figure.Figure)
使用示例:
from FScanpy import plot_prf_prediction
import matplotlib.pyplot as plt
# 1. 基本绘图
sequence = "ATGCGTACGTATGCGTACGTATGCGTACGT"
results, fig = plot_prf_prediction(sequence)
plt.show()
# 2. 自定义阈值和权重
results, fig = plot_prf_prediction(
sequence,
short_threshold=0.7, # 更高显示阈值
long_threshold=0.85, # 更高显示阈值
ensemble_weight=0.3, # 3:7 权重比例
title="自定义分析结果",
save_path="analysis.png",
figsize=(15, 10),
dpi=150
)
# 3. 高分辨率分析
results, fig = plot_prf_prediction(
sequence,
window_size=1, # 扫描每个位置
ensemble_weight=0.5, # 等权重
dpi=600 # 高分辨率
)
2. PRFPredictor 类方法
2.1 类初始化
from FScanpy import PRFPredictor
# 使用默认模型初始化
predictor = PRFPredictor()
# 使用自定义模型目录初始化
predictor = PRFPredictor(model_dir='/path/to/models')
2.2 predict_sequence() - 滑动窗口预测
方法签名:
def predict_sequence(self, sequence, window_size=3, short_threshold=0.1, ensemble_weight=0.4)
参数:
sequence:输入 DNA 序列window_size:滑动窗口大小(默认:3)short_threshold:短模型概率阈值(默认:0.1)ensemble_weight:集成中短模型权重(默认:0.4)
用法:
predictor = PRFPredictor()
results = predictor.predict_sequence(
sequence="ATGCGTACGT...",
window_size=1,
short_threshold=0.15,
ensemble_weight=0.35
)
2.3 predict_regions() - 基于区域的预测
方法签名:
def predict_regions(self, sequences, short_threshold=0.1, ensemble_weight=0.4)
参数:
sequences:399bp 序列的列表或 Seriesshort_threshold:短模型概率阈值(默认:0.1)ensemble_weight:集成中短模型权重(默认:0.4)
用法:
predictor = PRFPredictor()
sequences = ["ATGCGT...", "GCTATAG..."] # 399bp 序列
results = predictor.predict_regions(
sequences=sequences,
short_threshold=0.1,
ensemble_weight=0.4
)
2.4 predict_single_position() - 单位置预测
方法签名:
def predict_single_position(self, fs_period, full_seq, short_threshold=0.1, ensemble_weight=0.4)
参数:
fs_period:移码位点周围的 33bp 序列full_seq:长模型使用的 399bp 序列short_threshold:短模型概率阈值(默认:0.1)ensemble_weight:集成中短模型权重(默认:0.4)
用法:
predictor = PRFPredictor()
result = predictor.predict_single_position(
fs_period="ATGCGTACGTATGCGTACGTATGCGTACGTA", # 33bp
full_seq="ATGCGT..." * 133, # 399bp
short_threshold=0.1,
ensemble_weight=0.4
)
2.5 plot_sequence_prediction() - 类方法绘图
方法签名:
def plot_sequence_prediction(self, sequence, window_size=3, short_threshold=0.65,
long_threshold=0.8, ensemble_weight=0.4, title=None,
save_path=None, figsize=(12, 8), dpi=300)
用法:
predictor = PRFPredictor()
results, fig = predictor.plot_sequence_prediction(
sequence="ATGCGTACGT...",
window_size=3,
ensemble_weight=0.4
)
3. 实用函数
3.1 fscanr() - 从 BLASTX 检测 PRF 位点
函数签名:
def fscanr(
blastx_output: pd.DataFrame,
mismatch_cutoff: float = 10,
evalue_cutoff: float = 1e-5,
frameDist_cutoff: float = 10
) -> pd.DataFrame
参数:
blastx_output:包含必需列的 BLASTX 输出 DataFrame:qseqid,sseqid,pident,length,mismatch,gapopenqstart,qend,sstart,send,evalue,bitscore,qframe,sframe
mismatch_cutoff:最大允许错配数(默认:10)evalue_cutoff:E 值阈值(默认:1e-5)frameDist_cutoff:框架距离阈值(默认:10)
返回值:
pd.DataFrame:包含以下列的 PRF 位点:DNA_seqid:序列标识符FS_start,FS_end:移码开始和结束位置Pep_seqid:肽序列标识符Pep_FS_start,Pep_FS_end:肽移码位置FS_type:移码类型(-2, -1, 1, 2)Strand:链方向(+, -)
用法:
from FScanpy.utils import fscanr
import pandas as pd
# 加载 BLASTX 结果
blastx_data = pd.read_excel('blastx_results.xlsx')
# 检测 PRF 位点
prf_sites = fscanr(
blastx_output=blastx_data,
mismatch_cutoff=5, # 更严格的错配过滤
evalue_cutoff=1e-6, # 更严格的 E 值过滤
frameDist_cutoff=15 # 允许更大的框架距离
)
3.2 extract_prf_regions() - 提取 PRF 位点周围的序列
函数签名:
def extract_prf_regions(mrna_file: str, prf_data: pd.DataFrame) -> pd.DataFrame
参数:
mrna_file:mRNA 序列文件路径(FASTA 格式)prf_data:来自fscanr()输出的 DataFrame
返回值:
pd.DataFrame:提取的序列,包含以下列:DNA_seqid:序列标识符FS_start,FS_end:移码位置Strand:链方向399bp:提取的 399bp 序列FS_type:移码类型
用法:
from FScanpy.utils import extract_prf_regions
# 提取 PRF 位点周围的序列
prf_sequences = extract_prf_regions(
mrna_file='sequences.fasta',
prf_data=prf_sites
)
# 预测 PRF 概率
predictor = PRFPredictor()
results = predictor.predict_regions(prf_sequences['399bp'])
4. 数据访问函数
4.1 测试数据访问
from FScanpy.data import get_test_data_path, list_test_data
# 列出可用的测试数据
list_test_data()
# 获取测试数据路径
blastx_file = get_test_data_path('blastx_example.xlsx')
mrna_file = get_test_data_path('mrna_example.fasta')
region_file = get_test_data_path('region_example.csv')
seq_file = get_test_data_path('full_seq.xlsx')
完整工作流程示例
工作流程 1:完整序列分析
from FScanpy import predict_prf, plot_prf_prediction
import matplotlib.pyplot as plt
# 定义序列
full_seq = pd.read.excel(seq_file)
# 方法 1:简单预测
results = predict_prf(sequence=full_seq[0]['full_seq'])
print(f"发现 {len(results)} 个潜在位点")
# 方法 2:带可视化的预测
results, fig = plot_prf_prediction(
sequence=full_seq[0]['full_seq'],
window_size=1, # 扫描每个位置
short_threshold=0.3, # 显示概率 > 0.3 的位点
long_threshold=0.4, # 显示概率 > 0.4 的位点
ensemble_weight=0.4, # 4:6 权重比例
title="PRF 分析结果",
save_path="prf_analysis.png"
)
plt.show()
# 分析顶级预测
top_sites = results.nlargest(5, 'Ensemble_Probability')
print("前 5 个预测位点:")
for _, site in top_sites.iterrows():
print(f"位置 {site['Position']}: {site['Ensemble_Probability']:.3f}")
工作流程 2:基于区域的预测
from FScanpy import predict_prf
import pandas as pd
# 准备区域数据
region_data = pd.DataFrame({
'sample_id': ['sample1', 'sample2', 'sample3'],
'Long_Sequence': [
'ATGCGT...', # 399bp 序列 1
'GCTATAG...', # 399bp 序列 2
'TTACGGA...' # 399bp 序列 3
],
'known_label': [1, 0, 1] # 可选:用于验证的已知标签
})
# 预测 PRF 概率
results = predict_prf(
data=region_data,
ensemble_weight=0.3 # 偏向长模型(3:7 比例)
)
# 评估结果
if 'known_label' in results.columns:
threshold = 0.5
predictions = (results['Ensemble_Probability'] > threshold).astype(int)
accuracy = (predictions == results['known_label']).mean()
print(f"阈值 {threshold} 下的准确率:{accuracy:.3f}")
工作流程 3:基于 BLASTX 的分析流程
from FScanpy import PRFPredictor, predict_prf
from FScanpy.data import get_test_data_path
from FScanpy.utils import fscanr, extract_prf_regions
import pandas as pd
# 步骤 1:加载 BLASTX 数据
blastx_data = pd.read_excel(get_test_data_path('blastx_example.xlsx'))
print(f"加载了 {len(blastx_data)} 个 BLASTX 命中")
# 步骤 2:使用 FScanR 检测 PRF 位点
prf_sites = fscanr(
blastx_output=blastx_data,
mismatch_cutoff=10,
evalue_cutoff=1e-5,
frameDist_cutoff=10
)
print(f"检测到 {len(prf_sites)} 个潜在 PRF 位点")
# 步骤 3:提取 PRF 位点周围的序列
mrna_file = get_test_data_path('mrna_example.fasta')
prf_sequences = extract_prf_regions(
mrna_file=mrna_file,
prf_data=prf_sites
)
print(f"提取了 {len(prf_sequences)} 个序列")
# 步骤 4:预测 PRF 概率
predictor = PRFPredictor()
results = predictor.predict_regions(
sequences=prf_sequences['399bp'],
ensemble_weight=0.4
)
# 步骤 5:将结果与元数据结合
final_results = pd.concat([
prf_sequences.reset_index(drop=True),
results.reset_index(drop=True)
], axis=1)
# 步骤 6:分析结果
high_prob_sites = final_results[
final_results['Ensemble_Probability'] > 0.7
]
print(f"高概率 PRF 位点:{len(high_prob_sites)}")
# 显示顶级结果
print("\n顶级 PRF 预测:")
top_results = final_results.nlargest(3, 'Ensemble_Probability')
for _, row in top_results.iterrows():
print(f"序列 {row['DNA_seqid']}: {row['Ensemble_Probability']:.3f}")
工作流程 4:多序列自定义分析
from FScanpy import predict_prf, plot_prf_prediction
import matplotlib.pyplot as plt
# 多序列分析
sequences = [
"ATGCGTACGTATGCGTACGTATGCGTACGTAAGCCCTTTGAACCCAAAGGG",
"GCTATAGCATGCTATAGCATGCTATAGCATGCTATAGCATGCTATAGCAT",
"TTACGGATTACGGATTACGGATTACGGATTACGGATTACGGATTACGGAT"
]
# 批量预测
results = predict_prf(
sequence=sequences,
window_size=2,
ensemble_weight=0.5 # 等权重
)
# 按序列分析
for seq_id in results['Sequence_ID'].unique():
seq_results = results[results['Sequence_ID'] == seq_id]
max_prob = seq_results['Ensemble_Probability'].max()
print(f"{seq_id}: 最大概率 = {max_prob:.3f}")
# 可视化第一个序列
first_seq_results, fig = plot_prf_prediction(
sequence=sequences[0],
ensemble_weight=0.5,
title="第一个序列分析"
)
plt.show()
参数优化指南
1. 集成权重配置详解
1.1 模型互补优势原理
FScanpy 集成了两个具有互补优势的机器学习模型:
-
HistGB 模型(短模型):
- 擅长识别真阴性样本(正确排除非 PRF 位点)
- 基于 33bp 局部序列特征
- 预测更保守,假阳性率较低
- 适合高特异性要求的场景
-
BiLSTM-CNN 模型(长模型):
- 擅长识别真阳性样本(正确识别 PRF 位点)
- 基于 399bp 长距离序列特征
- 预测更敏感,能捕获更多潜在位点
- 适合高敏感性要求的场景
1.2 最优权重比例(4:6)
通过大量测试分析,我们确定了最优权重分布为 HistGB:BiLSTM-CNN = 4:6(ensemble_weight = 0.4):
- 最高 AUC 性能:在测试集上达到最佳的曲线下面积
- 平衡预测性能:在敏感性和特异性之间取得最佳平衡
- 降低极端错误:减少 BiLSTM-CNN 模型的过度预测风险
- 避免保守偏向:防止 HistGB 模型过于保守导致的模糊性
1.3 权重选择策略
根据研究目标选择合适的权重配置:
高通量筛选场景(偏向敏感性):
- 权重配置:
ensemble_weight = 0.6-0.8 - 适用场景:初步筛选、候选位点发现、探索性研究
- 优势:筛选更多候选位点,降低漏检风险
- 权衡:可能产生更多假阳性,需要后续验证
# 高敏感性配置示例
results = predict_prf(
sequence=sequence,
ensemble_weight=0.7, # 偏向 BiLSTM-CNN
short_threshold=0.05 # 降低短模型阈值
)
精确验证场景(偏向特异性):
- 权重配置:
ensemble_weight = 0.2-0.3 - 适用场景:候选验证、临床应用、高置信度预测
- 优势:减少假阳性,提高预测可靠性
- 权衡:可能遗漏部分真阳性位点
# 高特异性配置示例
results = predict_prf(
sequence=sequence,
ensemble_weight=0.25, # 偏向 HistGB
short_threshold=0.2 # 提高短模型阈值
)
平衡分析场景(推荐默认):
- 权重配置:
ensemble_weight = 0.4-0.6 - 适用场景:常规分析、综合评估、标准研究
- 优势:在敏感性和特异性间取得最佳平衡
- 推荐:大多数研究场景的首选配置
# 平衡配置示例
results = predict_prf(
sequence=sequence,
ensemble_weight=0.4, # 最优平衡比例
short_threshold=0.1 # 标准阈值
)
2. 阈值选择
- 短阈值:通常 0.1-0.3,控制计算效率
- 显示阈值:0.3-0.8,控制可视化显示
- 分类阈值:0.5(标准),根据验证数据调整
3. 窗口大小选择
- 精细分析:
window_size = 1(每个位置) - 标准分析:
window_size = 3(每第 3 个位置,默认) - 粗略分析:
window_size = 6-9(更快,细节较少)
故障排除
常见问题和解决方案
-
模型加载错误
# 检查模型目录 import FScanpy predictor = PRFPredictor(model_dir='/custom/path') -
大序列内存问题
# 使用更大的窗口大小减少计算负载 results = predict_prf(sequence=large_seq, window_size=9) -
可视化问题
# 调整图形参数 results, fig = plot_prf_prediction( sequence=seq, figsize=(20, 10), # 更大的图形 dpi=150 # 更低的分辨率 ) -
输入格式问题
# 确保正确的 DataFrame 格式 data = pd.DataFrame({ 'Long_Sequence': sequences, # 使用 'Long_Sequence' 或 '399bp' 'sample_id': ids })
性能优化
1. 批处理
# 高效处理多个序列
sequences = ["seq1", "seq2", "seq3", ...]
results = predict_prf(sequence=sequences, window_size=3)
2. 阈值优化
# 使用适当的 short_threshold 跳过不必要的长模型调用
results = predict_prf(
sequence=sequence,
short_threshold=0.2 # 更高阈值 = 更快处理
)
3. 内存管理
# 对于非常大的数据集,分块处理
chunk_size = 100
for i in range(0, len(large_dataset), chunk_size):
chunk = large_dataset[i:i+chunk_size]
chunk_results = predict_prf(data=chunk)
# 处理 chunk_results
引用
如果您使用 FScanpy,请引用我们的论文:[论文链接]