6.7 KiB
Abstract
FScanpy is a Python package designed to predict Programmed Ribosomal Frameshifting (PRF) sites in DNA sequences. This package integrates machine learning models, sequence feature analysis, and visualization capabilities to help researchers rapidly locate potential PRF sites.
Introduction
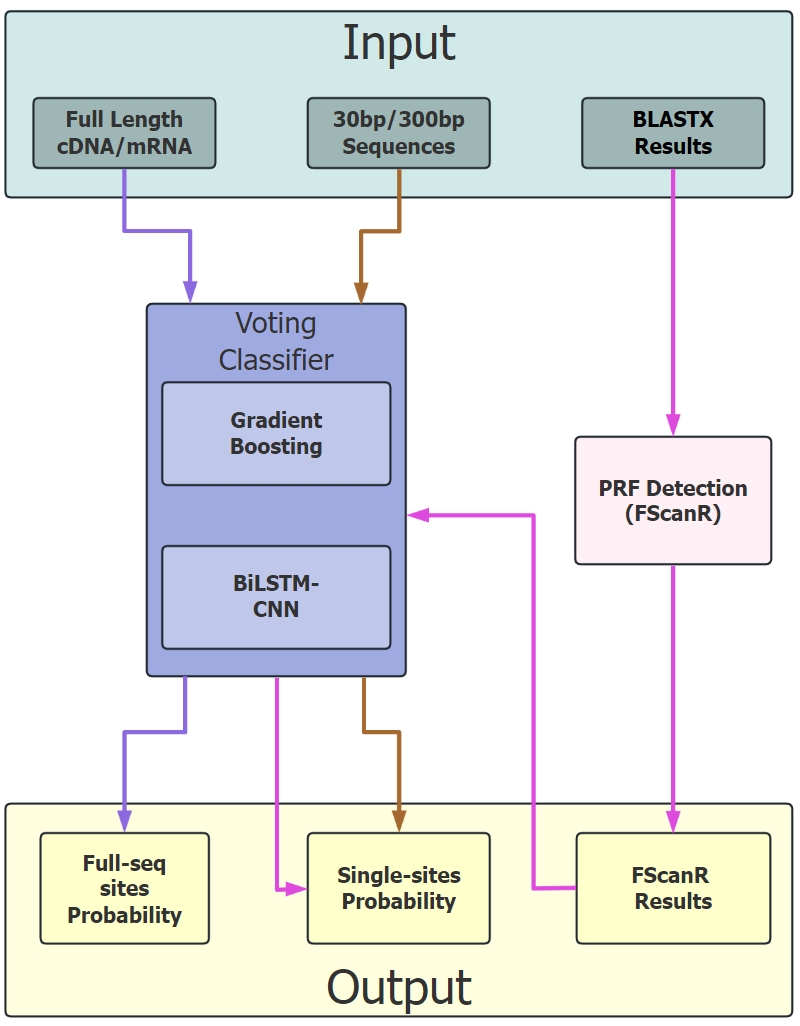
FScanpy is a Python package dedicated to predicting Programmed Ribosomal Frameshifting (PRF) sites in DNA sequences. It integrates machine learning models (Gradient Boosting and BiLSTM-CNN) along with the FScanR package to furnish precise PRF predictions. Users are capable of employing three types of data as input: the entire cDNA/mRNA sequence that requires prediction, the nucleotide sequence in the vicinity of the suspected frameshift site, and the peptide library blastx results of the species or related species. It anticipates the input sequence to be in the + strand and can be integrated with FScanR to augment the accuracy.
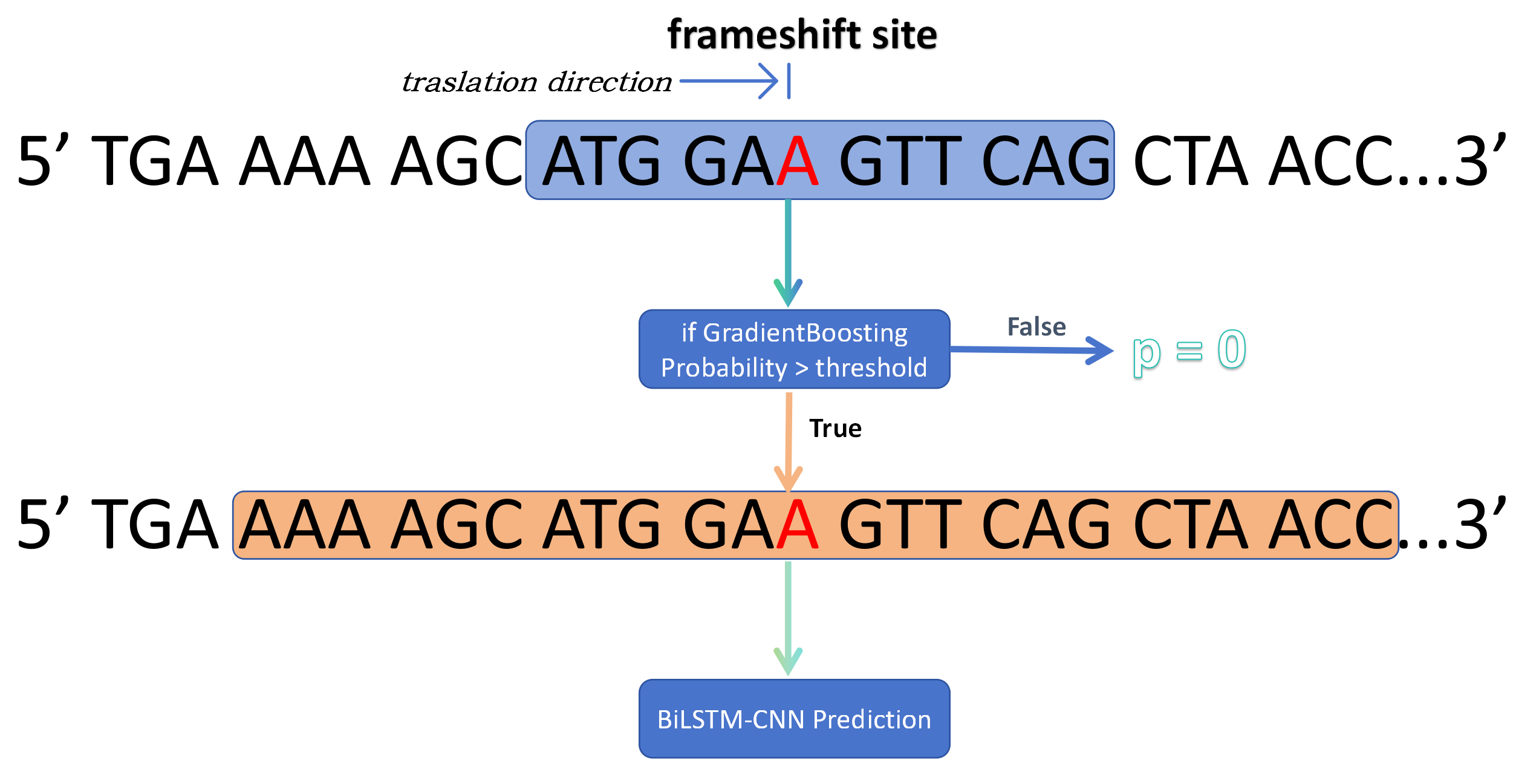 For the prediction of the entire sequence, FScanpy adopts a sliding window approach to scan the entire sequence and predict the PRF sites. For regional prediction, it is based on the 33-bp and 399-bp sequences in the 0 reading frame around the suspected frameshift site. Initially, the Short model (HistGradientBoosting) will predict the potential PRF sites within the scanning window. If the predicted probability exceeds the threshold, the Long model (BiLSTM-CNN) will predict the PRF sites in the 399bp sequence. Then, ensemble weighting combines the two models to make the final prediction.
For the prediction of the entire sequence, FScanpy adopts a sliding window approach to scan the entire sequence and predict the PRF sites. For regional prediction, it is based on the 33-bp and 399-bp sequences in the 0 reading frame around the suspected frameshift site. Initially, the Short model (HistGradientBoosting) will predict the potential PRF sites within the scanning window. If the predicted probability exceeds the threshold, the Long model (BiLSTM-CNN) will predict the PRF sites in the 399bp sequence. Then, ensemble weighting combines the two models to make the final prediction.
For PRF detection from BLASTX output, FScanR identifies potential PRF sites from BLASTX alignment results, acquires the two hits of the same query sequence, and then utilizes frameDist_cutoff, mismatch_cutoff, and evalue_cutoff to filter the hits. Finally, FScanpy is utilized to predict the probability of PRF sites.
Background
Ribosomal frameshifting, also known as translational frameshifting or translational recoding, is a biological phenomenon that occurs during translation that results in the production of multiple, unique proteins from a single mRNA. The process can be programmed by the nucleotide sequence of the mRNA and is sometimes affected by the secondary, 3-dimensional mRNA structure. It has been described mainly in viruses (especially retroviruses), retrotransposons and bacterial insertion elements, and also in some cellular genes.
Key features of FScanpy include:
- Integration of two predictive models:
- Short Model (HistGradientBoosting): Analyzes local sequence features centered around potential frameshift sites (33bp).
- Long Model (BiLSTM-CNN): Analyzes broader sequence features (399bp).
- Supports PRF prediction across various species.
- Can be combined with FScanR for enhanced accuracy.
Installation (python>=3.7)
1. Use pip
pip install FScanpy
2. Clone from GitHub
git clone https://github.com/.../FScanpy.git
cd your_project_directory
pip install -e .
Methods and Usage
1. Load model and test data
from FScanpy import PRFPredictor
from FScanpy.data import get_test_data_path, list_test_data
predictor = PRFPredictor() # load model
list_test_data() # list all the test data
blastx_file = get_test_data_path('blastx_example.xlsx')
mrna_file = get_test_data_path('mrna_example.fasta')
region_example = get_test_data_path('region_example.xlsx')
2. Predict PRF Sites in a Full Sequence
Use the predict_sequence() method to scan the entire sequence:
results = predictor.predict_sequence(
sequence='ATGCGTACGTATGCGTACGTATGCGTACGT',
window_size=3, # Scanning window size
short_threshold=0.1, # Short model threshold
ensemble_weight=0.4 # Ensemble weight (Short:Long = 0.4:0.6)
)
# With visualization
results, fig = predictor.plot_sequence_prediction(
sequence='ATGCGTACGTATGCGTACGTATGCGTACGT',
ensemble_weight=0.4
)
3. Predict PRF in Specific Regions
Use the predict_regions() method to predict PRF in known regions of interest:
import pandas as pd
region_example = pd.read_excel(get_test_data_path('region_example.xlsx'))
results = predictor.predict_regions(
sequences=region_example['399bp'],
ensemble_weight=0.4
)
4. Identify PRF Sites from BLASTX Output
BLASTX Output should contain the following columns: qseqid, sseqid, pident, length, mismatch, gapopen, qstart, qend, sstart, send, evalue, bitscore, qframe, sframe.
Use the FScanR function to identify potential PRF sites from BLASTX alignment results:
from FScanpy.utils import fscanr
blastx_output = pd.read_excel(get_test_data_path('blastx_example.xlsx'))
fscanr_result = fscanr(blastx_output,
mismatch_cutoff=10, # Allowed mismatches
evalue_cutoff=1e-5, # E-value threshold
frameDist_cutoff=10) # Frame distance threshold
5. Extract PRF Sites and Evaluate
Use the extract_prf_regions() method to extract PRF site sequences from mRNA sequences:
from FScanpy.utils import extract_prf_regions
prf_regions = extract_prf_regions(
mrna_file=get_test_data_path('mrna_example.fasta'),
prf_data=fscanr_result
)
prf_results = predictor.predict_regions(prf_regions['399bp'])
Complete Workflow Example
from FScanpy import PRFPredictor, predict_prf, plot_prf_prediction
from FScanpy.data import get_test_data_path, list_test_data
from FScanpy.utils import fscanr, extract_prf_regions
import pandas as pd
# Initialize predictor
predictor = PRFPredictor()
# Method 1: Sequence prediction
sequence = 'ATGCGTACGTATGCGTACGTATGCGTACGT'
results = predict_prf(sequence=sequence, ensemble_weight=0.4)
# Method 2: Region prediction
region_data = pd.read_excel(get_test_data_path('region_example.xlsx'))
results = predict_prf(data=region_data, ensemble_weight=0.4)
# Method 3: BLASTX pipeline
blastx_output = pd.read_excel(get_test_data_path('blastx_example.xlsx'))
fscanr_result = fscanr(blastx_output, mismatch_cutoff=10, evalue_cutoff=1e-5, frameDist_cutoff=10)
prf_regions = extract_prf_regions(get_test_data_path('mrna_example.fasta'), fscanr_result)
prf_results = predictor.predict_regions(prf_regions['399bp'])
# Visualization
results, fig = plot_prf_prediction(sequence, ensemble_weight=0.4, save_path='prediction.png')
Citation
If you use FScanpy, please cite our paper: [Paper Link]